读取浮雕对齐文件时,AlignIO会发出'AssertionError'
我已经坚持了三天的问题......到处搜索,发布在Biostar上,仍在等待EMBL回复电子邮件......如果我有更多的代表,我会获得赏金。
使用EMBOSSwin needle()(成对全局比对)对齐序列后,我得到pair格式的对齐文件,文件扩展名为.needle。我想使用Biopython来读取这些对齐以供以后分析。
我按照Biopython's AlignIO wiki中的说明使用AlignIO.read(open('alignment.needle'),'emboss'),但我不断获得AssertionError。
我的代码:
>>> from Bio import AlignIO
>>> alignment = AlignIO.read(open("data/all/out/pair1_alignment.needle"), "emboss")
我的错误:
Traceback (most recent call last):
File "<console>", line 1, in <module>
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 423, in read
first = next(iterator)
File "C:\Python27\lib\Bio\AlignIO\__init__.py", line 370, in parse
for a in i:
File "C:\Python27\lib\Bio\AlignIO\EmbossIO.py", line 150, in __next__
assert seq.replace("-", "") != ""
AssertionError
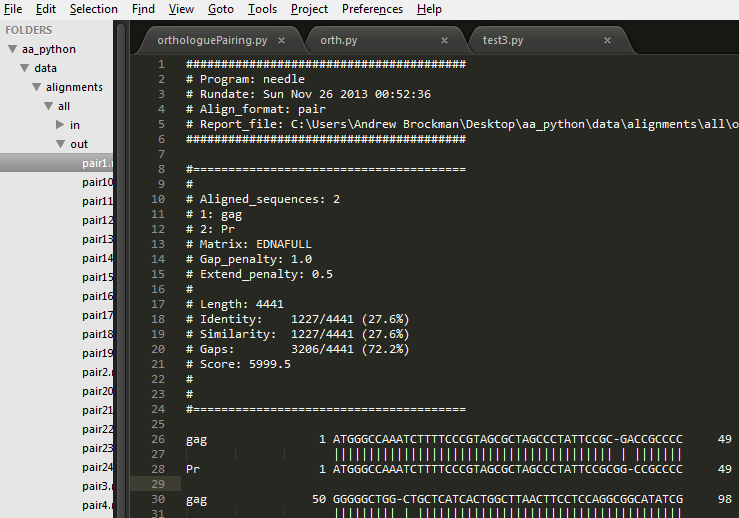
示例对齐文件:
下载对齐文件here
版本:
- Windows 7
- Python版本2.7.3
- Biopython版本1.63
- EMBOSS版本2.10.0-0.8
线索:
我怀疑这可能与我在实际进行对齐时遇到的警告信息有关,这是由EMBOSS needle()函数输出的:
Warning: Sequence character string not found in ajSeqCvtKS
2 个答案:
答案 0 :(得分:2)
BioStars上的重复帖子,http://www.biostars.org/p/87226/#87399
这似乎是EMBOSS输出的微妙变化。你有一个非常老的版本,EMBOSS版本2。10。0(2005年2月),你的输出文件有这样的行:
gag 1288 -------------------------------------------------- 1287
使用较新版本的EMBOSS(例如6.3.0),给出如下行:
gag 1287 -------------------------------------------------- 1287
Biopython解析器期望后者用于没有字母的对齐部分(例如,当一个序列比另一个序列长得多时),其中开始和结束坐标一致。请更新您的EMBOSS副本,然后解析器应该很高兴。目前的EMBOSS版本是6.5.0版本。
答案 1 :(得分:1)
问题是您将错误的格式文件传递给Biopython。解释如下。
格式
您链接到的文件格式为srspair(请参阅pair1_aligned.fasta标题)。值得注意的是,不是 FASTA format - 这是一种完全不同的格式。
深入研究source of Biopython's EmbossIO,我们可以看到EmbossIterator(格式为AlignIO.read时由'emboss'调用)仅用于处理格式{ {1}}和pair(有关各种格式的说明,请参阅Alignment formats)。
解决方案
如果您以simple格式导出EMBOSS的输出(然后像之前一样调用pair),那么这应该可以解决您的问题。
- 读取浮雕对齐文件时,AlignIO会发出'AssertionError'
- 浮雕针()警告:“在ajSeqCvtKS中找不到序列字符”......?
- 简单的布尔索引给出了AssertionError:无法创建BlockManager
- 模拟私有void方法会产生AssertionError
- BioPython AlignIO ValueError说字符串长度必须相同?
- 读取excel时带有pandas的AssertionError
- Mockito:AssertionError期望True但是给出False
- BioPython AlignIO序列长度必须相同[多个文件]
- 来自biopython AlignIO的多个输入文件作为单个文件输出
- 我写了这段代码,但我无法理解我的错误
- 我无法从一个代码实例的列表中删除 None 值,但我可以在另一个实例中。为什么它适用于一个细分市场而不适用于另一个细分市场?
- 是否有可能使 loadstring 不可能等于打印?卢阿
- java中的random.expovariate()
- Appscript 通过会议在 Google 日历中发送电子邮件和创建活动
- 为什么我的 Onclick 箭头功能在 React 中不起作用?
- 在此代码中是否有使用“this”的替代方法?
- 在 SQL Server 和 PostgreSQL 上查询,我如何从第一个表获得第二个表的可视化
- 每千个数字得到
- 更新了城市边界 KML 文件的来源?